D. Nemcsok, A. Kovács, K.
Mészáros-Szécsényi, V. M. Leovac
Vibrational
spectroscopic and theoretical study of 3,5-dimethyl-1-thiocarboxamide pyrazole
(L) and the complexes Co2L2Cl4, Cu2L2Cl4
and Cu2L2Br2.
Chemical Physics, 328
(2006) 85-92
In the present paper we report a joint
experimental and theoretical study of 3,5-dimethyl-1-thiocarboxamide pyrazole
(L) and its complexes Co2L2Cl4, Cu2L2Cl4
and Cu2L2Br2. DFT computations were used to
model the structural and bonding properties of the title compounds as well as
to derive a reliable force field for the normal coordinate analysis of L. The
computations indicated the importance of hydrogen bonding interactions in
stabilising the global minimum structures on the potential energy surfaces. In
contrast to the S-bridged binuclear Cu2L2Br2
complex found in the crystal, our computations predicted the formation of
(CuLBr)2 dimers in the isolated state stabilized by very strong (53
kJ/mol) N–H…Br hydrogen bonding interactions. On the
basis of FT-IR and FT-Raman experiments and the DFT-derived scaled quantum
mechanical force field we carried out a complete normal coordinate analysis of
L. The FT-IR spectra of the three complexes were interpreted using the present
assignment of L, literature data and computed results.
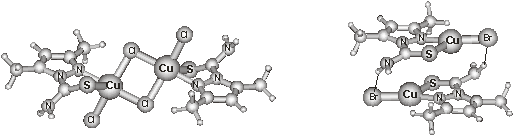
The optimised structures of Cu2L2Cl4 and
(CuLBr)2.