A.
Kovács, V. Izvekov, K. Zauer, K. Ohta
Strong intramolecular hydrogen bonding
and molecular vibrations of 9-hydroxyphenalen-1-one
The Journal of Physical Chemistry
A, 105 (2001) 5000-5009
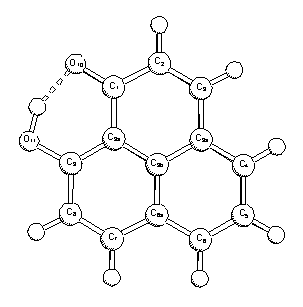
The strong intramolecular hydrogen bonding in
9-hydroxyphenalen-1-one (1) has been investigated by means of quantum
chemical calculations and vibrational spectroscopy. Both ab initio molecular
orbital (MP2) and density functional theory (B3-LYP, B3-P86) calculations
predict a double-minimum potential energy surface with a low (ca. 10 kJ/mol)
barrier. The hydrogen bonding energy, estimated by comparison with the
non-hydrogen-bonded anti conformer, is ca. 60 kJ/mol. Our comparative study of
the three theoretical levels revealed the very good performance of the B3-LYP
density functional in conjunction with a diffuse polarized valence triple-zeta
basis set while the B3-P86 functional tends to overestimate considerably the
strong hydrogen bonding interaction. Based on a joint analysis of the
energetics and molecular geometry, the interaction in 1 can be
classified as a border case between traditional and short-strong hydrogen
bonds. The charge distribution refers to a strong ionic character of the
hydrogen bonding interaction. The vibrational properties of the molecule have
been investigated by a combined experimental (FT-IR, FT-Raman) and theoretical
analysis. The deficiencies of the computed harmonic force field were corrected
by the scaled quantum mechanical (SQM) method of Pulay et al. As a result of
our SQM analysis, 45 from a total of 63 fundamentals of the molecule were
assigned with an rms deviation of 6.9 cm-1 between the experimental
and scaled frequencies. The most characteristic effect of hydrogen bonding on
the vibrational properties of 1 is the enhanced mixing of the CO and OH
vibrations with each other and with the skeletal modes.