Attila Kovács, Andrea Szabó,
Dénes Nemcsok, and István Hargittai
Blue-shifting
C–H…X (X = O, halogen) hydrogen bonds in the dimers of formaldehyde
derivatives
The Journal of Physical Chemistry A, 106 (2002) 5671-5678.
The potential energy surface (PES) of the
dimers of formaldehyde derivatives CH(O)Y (Y = H, CH3, F, Cl, Br, I)
has been investigated by means of quantum chemical calculations at the
MP2/6-311++G** level. Several minima have been found on the PES characterized
by various combinations of C–H…X (X = O, halogen) contacts. The
computed dimerization energies revealed the importance of dispersion forces in
the formation of [CH(O)Y]2
dimers, while only a lesser role of the intermolecular H…X interactions.
The most characteristic geometrical properties of the dimers are the H…X distances
being near the sum of the van der Waals radii of the contacting atoms, the
lengthening of the contacting C–X bonds, and the general shortening of
the C–H bonds by 0.001-0.004 Å with respect to the monomers. The
latter bond shortening is responsible for the characteristic blue shift of the
CH stretching frequencies in the dimers. A natural bond orbital (NBO) analysis
revealed a slight decrease in the population of the contacting s*CH orbitals and alterations in the intramolecular
charge transfer effects as the primary reason of the C–H contraction.
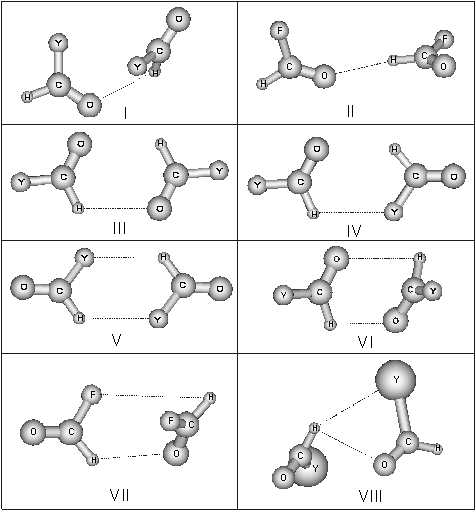
Optimized
structures of the possible dimers of formaldehyde derivatives