Attila Kovács and Rudy J.M. Konings
A theoretical
study of the structure and bonding of UOX4 (X=F, Cl, Br, I)
molecules.
The
importance of inverse trans influence.
ChemPhysChem, 7
(2006) 455-462
The structural and bonding properties of the
uranium(VI) oxyhalides UOX4 (X = F, Cl, Br, I) have been
investigated by quantum chemical calculations at three different levels of
theory: quasi-relativistic density functional theory (DFT) in conjunction with
a triple-zeta all electron basis set as well as MP2 and the
Becke3-Perdew-Wang91 exchange-correlation functional in conjunction with
relativistic effective core potentials (ECP). The computations located four
stationary points on the potential energy surface: a tetragonal pyramid with O in the apical position (C4v), a trigonal bipyramid with O in the equatorial
position (C2v), a trigonal
bipyramid with O in the axial position (C3v),
and a Cs structure derived
from C3v by opening the Xeq–U–Xeq
angle to near 180º. The Cs
minimum, however, seems to be an artifact of the Becke-Perdew functional on the
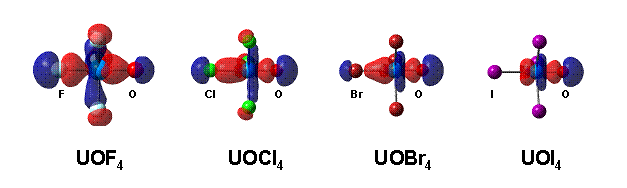
flat potential energy surface. In the C3v
structures the linear Xax–U=O moieties show clearly the
geometrical consequences of the inverse trans influence (ITI) effect. This
interaction can stabilise these sterically less favoured geometries. Two
important trends are revealed by our computations: (i) UOX4 with the
small X = F prefers the C3v
structure, whereas with increasing halogen size the sterically less crowded C2v structure gets more importance.
(ii) MP2 theory accounts to a less extent for the ITI effect with respect to
DFT, which results in different structural preferences (MP2 for C2v, DFT for C3v) in the heavier halides.
In addition, an important bonding property of the UOX4 molecules is
the clear triple-bond character of the formally double U=O bonds.
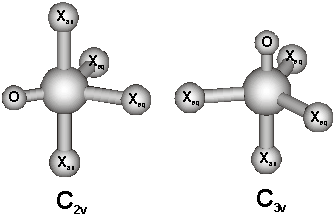
The relevant structures.
The A1 molecular orbital representing sU=O and sU-X contributions in the C3v
structures.
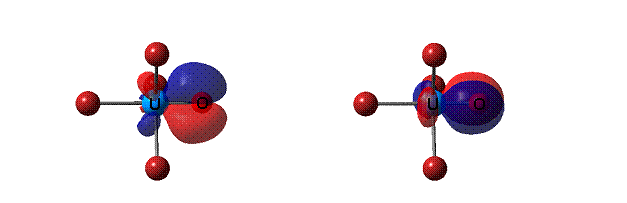
The two p orbitals of the UºO triple bond.