Attila Kovács
Theoretical
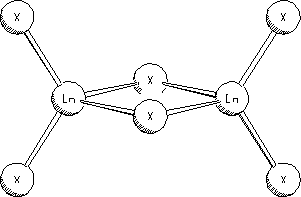
study of rare earth trihalide dimers Ln2X6
(Ln=La, Dy;
X=F, Cl, Br, I)
Chemical Physics Letters, 319 (2000) 238-246
The energetics, structure, bonding and molecular
vibrations of Ln2X6 (Ln = La, Dy; X = F, Cl, Br, I) dimers have been
studied using density functional theory in conjunction with polarised
triple-zeta valence basis sets and relativistic effective core potentials for
the heavy atoms. Selection of the
computational method was based on test calculations investigating the
convergence to basis set saturation.
Trends have been determined in the energy of dimer formation as well as
in the variation of the geometrical parameters. The ionic bonding in the dimers was
characterised by natural atomic charges as well as by a topological analysis of
the electron density distribution according to Bader’s theorem.